
ę╗Īó╗∙▒Šą┼Žó
Ė┼╩÷Ż║Ž╚╠ņąį╝ā╝t╝Ü(x©¼)░¹į┘╔·šŽĄKąįžÜč¬╩Ūę╗ĘN║╦╠Ū¾wĄ░░ūĮY(ji©”)śŗ(g©░u)╗∙ę“═╗ūāī¦(d©Żo)ų┬║╦╠Ū¾w╔·╬’║Ž│╔«É│ŻĄ─╔┘ęŖ(ji©żn)▀zé„ąį╝▓▓ĪŻ¼ėųĘQDiamond-Blackfan AnemiaŻ©DBAŻ®Ż¼╩Ūė╔╝t╝Ü(x©¼)░¹ā╚(n©©i)į┤ąį╔·│╔╚▒Ž▌╦∙ų┬Ż¼│╩│Ż╚Š╔½¾w’@ąį╗“ļ[ąį▀zé„ĪŻĮ^┤¾ČÓöĄ(sh©┤)╗╝ā║Ų▓Ī░l(f©Ī)╔·ė┌1Üqęįā╚(n©©i)Ż¼▒Ē¼F(xi©żn)×ķ┤¾╝Ü(x©¼)░¹ąįžÜč¬Īó╣Ū╦Ķ╝tŽĄ╝Ü(x©¼)░¹├„’@£p╔┘Īó░l(f©Ī)ė²╗¹ą╬║═─[┴÷ęūĖąąįį÷Ė▀Ą╚ĪŻ
▓Īę“Ż║DBAė┌1936─Ļė╔īW(xu©”)š▀Josephs╩ū┤╬ł¾(b©żo)Ą└Ż¼1938─Ļė╔īW(xu©”)š▀Diamond║═Blackfanł¾(b©żo)Ą└├Ķ╩÷Ż¼ę“Č°Ą├├¹ĪŻDBA╩Ūę╗ĘN║╦╠Ū¾w║Ž│╔šŽĄKąį╝▓▓ĪŻ¼╩Ūė░Ēæ║╦╠Ū¾w║Ž│╔Ą─╗∙ę“═╗ūā╦∙ų┬ĪŻ─┐Ū░šJ(r©©n)×ķ║╦╠Ū¾w╣”─▄╚▒Ž▌ę²Ų▀xō±ąį╝tŽĄ╔·│╔▓╗┴╝Ż¼▒Ē¼F(xi©żn)×ķ╝tŽĄČ©Ž“ūµ╝Ü(x©¼)░¹┤µį┌į÷ų│ĪóĘų╗»ĪóĄ“═÷╝░ī”(du©¼)╝Ü(x©¼)░¹ę“ūė¤o(w©▓)Ę┤æ¬(y©®ng)Ą─ā╚(n©©i)į┌╚▒Ž▌Ż¼ę“═¼Ģr(sh©¬)ė^▓ņĄĮDBA╗╝š▀ėąÅVĘ║Ą─▄|¾w╗¹ą╬Ż¼┐╔ęį│÷¼F(xi©żn)Ųõ╦¹č¬╝Ü(x©¼)░¹«É│ŻŻ¼░l(f©Ī)š╣×ķį┘╔·šŽĄKąįžÜ謯¼╠ß╩ŠDBAę▓┐╔└█╝░│²╝tŽĄįņč¬═ŌĄ─ČÓéĆ(g©©)ŽĄĮy(t©»ng)ĪŻ┴Ē═ŌŻ¼║╦╠Ū¾wĄ─║Ž│╔╩▄ōpę▓ė░Ēæ─[┴÷Ą░░ūp53Ż©tumor protein p53Ż¼TP53Ż®─[┴÷ęųųŲ═©┬ĘĄ─ĘĆ(w©¦n)Č©ąį║═╗ŅąįŻ¼▀@▒╗šJ(r©©n)×ķ╩Ū╝▓▓Ī┼R┤▓▒Ē¼F(xi©żn)Ż©░³└©╝t╝Ü(x©¼)░¹╔·│╔╩▄ōpĪó─[┴÷ęūĖąŻ®Ą─įŁę“Ż¼p53╝ż╗Ņ╝░Ž┬ė╬╩┬╝■ī¦(d©Żo)ų┬╝Ü(x©¼)░¹ų▄Ų┌Ę²½@║═╝Ü(x©¼)░¹Ą“═÷ĪŻ═©│Ż▒Ē¼F(xi©żn)×ķ│Ż╚Š╔½¾w’@ąį▀zé„Ż¼Ą½╩Ūį┌ę╗éĆ(g©©)╝ęūÕųąć└(y©ón)ųž│╠Č╚▓Ņ«É║▄┤¾Ż©═Ō’@┬╩ĮĄĄ═Ż®ĪŻ
═©▀^(gu©░)▀BµiĘų╬÷Įę╩ŠDBAĄ─▀zé„╗∙ę“╬╗³c(di©Żn)ęč▒╗Ļæ└m(x©┤)Öz│÷ĪŻ╝s25ŻźĄ─DBA╗╝š▀╩ŪŠÄ┤a║╦╠Ū¾wĄ░░ū19Ą─╗∙ę“Ż©RPS19Ż®░l(f©Ī)╔·═╗ūāĪŻęčėąŠÄ┤a║╦╠Ū¾w┤¾üå╗∙╗∙ę“Ż©RPL35AĪóRPL5ĪóRPL11Ż®║═║╦╠Ū¾wąĪüå╗∙╗∙ę“Ż©RPS24ĪóRPS17ĪóRPS7ĪóRPS10ĪóRPS26Ż®ų┬▓Īąį═╗ūāĄ─├Ķ╩÷Ż¼į┌50Żźęį╔ŽĄ─DBA╗╝š▀ųą│÷¼F(xi©żn)ĪŻŲõųąRPL5 ╗“ RPL11 ═╗ūā╗╝š▀Ė³│Ż│÷¼F(xi©żn)▄|¾w╗¹ą╬ĪŻ
DBA╗╝ā║EPO╔·│╔š²│ŻŻ¼╔ą¤o(w©▓)EPO┐╣¾wĄ─├Ķ╩÷ĪŻ2012─Ļ蹊┐░l(f©Ī)¼F(xi©żn)Ż¼DBA╗╝š▀Ą─įņč¬▐D(zhu©Żn)õøę“ūėGATA1ę▓┐╔│÷¼F(xi©żn)╝¶Įėģ^(q©▒)═╗ūāŻ¼▀@ĘN═╗ūā╩Ū│²║╦╠Ū¾wĄ░░ū«É│Żų«═Ō┴Ēę╗ĘN┐╔ī¦(d©Żo)ų┬DBAĄ─ę“╦žŻ¼┐╔ōp║”╚½ķL(zh©Żng)Ą░░ūŻ©ę╗ĘN╝tŽĄĘų╗»╦∙▒žąĶĄ─▐D(zhu©Żn)õøę“ūėŻ®Ą─╔·│╔Ż¼×ķX▀Bµi╗“│Ż╚Š╔½¾wļ[ąį▀zé„ĪŻŲõ╦¹Ą─ÖC(j©®)ųŲ┐╔─▄ėąįŁ░®╗∙ę“c-mycĄ─╝ż╗ŅĪŻ
┴„ąą▓ĪīW(xu©”)Ż║ė╔ė┌DBAŅH×ķ╔┘ęŖ(ji©żn)Ż¼Ųõ┤_ŪąĄ─░l(f©Ī)▓Ī┬╩ļyęį┤_Č©ĪŻÜWų▐╗žŅÖąį蹊┐▒Ē├„Ż¼DBAį┌Ī▄15Üqā║═»ųą─Ļ░l(f©Ī)▓Ī┬╩╝s×ķ1.5/100╚f(w©żn)Ī½5.0/100╚f(w©żn)ĪŻ┤╦▓Ī░l(f©Ī)╔·ė┌ŗļėūā║Ż¼ČÓöĄ(sh©┤)╗╝ā║│÷╔·║¾2ų▄ų┴2─Ļ░l(f©Ī)▓ĪŻ¼│¼▀^(gu©░)90Żź╗╝ā║į┌1Üqā╚(n©©i)┤_į\Ż¼Ųõųą35Żź╩Ūį┌│÷╔·1éĆ(g©©)į┬ā╚(n©©i)į\öÓĄ─ĪŻ╗╝ā║ČÓį┌Š═į\Ģr(sh©¬)ėą├„’@Ą─žÜč¬ĪŻ▒Š▓Ī─ą┼«╗╝š▀ų«▒╚╝s×ķ1.1Ī├1Ż¼10ŻźĪ½25Żźėą╝ęūÕ╩ĘĪŻ▒M╣▄┤¾ČÓöĄ(sh©┤)▓Ī└²ł¾(b©żo)Ą└×ķĖ▀╝ė╦„╚╦Ż¼Ą½╩Ū▀@ĘN▓Ī└²ęčį┌ČÓĘN▓╗═¼Ą─╚╦ĘNųą▒╗░l(f©Ī)¼F(xi©żn)ĪŻ
Č■Īó╝▓▓Īį\öÓ
┼R┤▓▒Ē¼F(xi©żn)Ż║
1.╣Ū╦Ķ╦źĮ▀
35Żź╗╝ā║│÷╔·Ģr(sh©¬)╝┤▒Ē¼F(xi©żn)×ķžÜ謯¼│Żė┌╔·║¾2ų▄ų┴2─Ļ┤_į\ĪŻžÜč¬×ķ┤¾╝Ü(x©¼)░¹╗“š²╝Ü(x©¼)░¹š²╔½╦žąįĪóŠW(w©Żng)┐Ś╝t╝Ü(x©¼)░¹£p╔┘Īó░ū╝Ü(x©¼)░¹š²│Ż╗“▌pČ╚ĮĄĄ═Īó謹Ī░Õš²│Ż╗“▌pČ╚į÷Ė▀Īó╣Ū╦Ķ╝tŽĄį÷╔·Ą═Ž┬Č°┴ŻŽĄ║═Š▐║╦╝Ü(x©¼)░¹ŽĄį÷╔·╗Ņ▄SĪŻ
2.Ž╚╠ņ░l(f©Ī)ė²«É│Ż
30ŻźĪ½50ŻźDBA╗╝š▀┐╔─▄┤µį┌Ž╚╠ņ░l(f©Ī)ė²«É│ŻŻ¼ų„ę¬╔µ╝░Ņ^▓┐Īó╔Žų½Īóą─┼K║═├┌─“╔·ų│ŽĄĮy(t©»ng)ĪŻ╚ń╔Ē▓─░½ąĪŻ¼’B├µ▓┐╗¹ą╬░³└©’B├µ▓┐▀^(gu©░)īÆĪóīÆŲĮ▒Ū┴║ĪóąĪŅ^ĪóŽ╚╠ņąį░ūā╚(n©©i)šŽĪóŪÓ╣Ōč█Īóą▒ęĢĪóė▓ļ±Ė▀╣░╝░┤Įļ±┴č╔§ų┴╠ž╝{ŠC║Žš„═Ō├▓Ż¼─┤ųĖ╗¹ą╬Ż¼Ž╚╠ņąįą─č¬╣▄░l(f©Ī)ė²«É│ŻŻ¼├┌─“╔·ų│Ų„╣┘╗¹ą╬ĪŻ┤µį┌╔Ē¾wĄ─«É│Ż▓ó▓╗─▄ŅA(y©┤)£y(c©©)╗╝š▀č¬ę║ŽĄĮy(t©»ng)╝▓▓ĪĄ─ć└(y©ón)ųž│╠Č╚ĪŻ30Żź╩▄└█╗╝š▀┐╔│÷¼F(xi©żn)╔·║¾Ą─╔·ķL(zh©Żng)▀tŠÅŻ¼═©│Ż░ķėąŲõ╦¹Ž╚╠ņ╗¹ą╬ĪŻ╚╗Č°Ż¼RPS19 ╗∙ę“▀Bµi═╗ūāät▓╗Ģ■(hu©¼)░ķėą╔·ķL(zh©Żng)▀tŠÅĪŻ
3.░®░YęūĖąąįį÷╝ė
DBA╗╝š▀─[┴÷░l(f©Ī)╔·┬╩╝s×ķ4ŻźŻ¼Ė▀ė┌═¼─Ļ²gš²│Ż╚╦╚║ĪŻŪę─[┴÷░l(f©Ī)╔·─Ļ²gįńŻ¼ųą╬╗─Ļ²g×ķ15ÜqĪŻ░³└©č¬ę║ŽĄĮy(t©»ng)╝▒ąį░ūč¬▓ĪĪó╣Ū╦Ķį÷╔·«É│ŻŠC║Žš„Īó┴▄░═┴÷╝░īŹ(sh©¬)¾w┴÷╚ń╣Ū╚Ō┴÷Ī󹞎┘░®ĪóĖ╬╝Ü(x©¼)░¹░®Īó║┌╔½╦ž┴÷Īó└wŠSĮM┐Ś╝Ü(x©¼)░¹┴÷Īó╬Ė░®║═─c░®Ą╚ĪŻ╗╝─[┴÷║¾,▌^╗╝═¼śė─[┴÷Ą─ę╗░Ń╚╦╚║ŅA(y©┤)║¾▓ŅŻ¼▓Ī╦└┬╩Ė▀ĪŻ
▌oų·Öz▓ķŻ║
1.č¬│ŻęÄ(gu©®)╝░ŠW(w©Żng)┐Ś╝t╝Ü(x©¼)░¹ėŗ(j©¼)öĄ(sh©┤)Öz£y(c©©)
ī”(du©¼)ė┌ę╔╦ŲDBAš▀æ¬(y©®ng)▀M(j©¼n)ąąŻ¼ę╗░Ń╩Ū┤¾╝Ü(x©¼)░¹ąįžÜ謯¼╝t╝Ü(x©¼)░¹ŲĮŠ∙╚▌ĘeŻ©mean corpuscular volumeŻ¼MCVŻ®į÷┤¾ĪŻ═©│Żį┌į\öÓĢr(sh©¬)žÜč¬║▄ć└(y©ón)ųžŻ¼2éĆ(g©©)į┬ęįŽ┬Ą─ŗļā║ŲĮŠ∙č¬╝tĄ░░ū╦«ŲĮ65g/LŻ¼2éĆ(g©©)į┬ęį╔ŽĄ─ŗļā║×ķ40g/LĪŻ╚╗Č°Ż¼ę╗ą®ĘŪĄõą═Ą─DBA╗╝š▀āHėą▌pČ╚žÜ謯¼╗“š▀āHėą▌p╬óĄ─╝tŽĄ«É│ŻĪŻŠW(w©Żng)┐Ś╝t╝Ü(x©¼)░¹ėŗ(j©¼)öĄ(sh©┤)├„’@ĮĄĄ═ĪŻ═©│Ż╗╝š▀Ą─░ū╝Ü(x©¼)░¹║═謹Ī░Õėŗ(j©¼)öĄ(sh©┤)▓╗╩▄ė░ĒæĪŻ
2.č¬╝tĄ░░ūļŖėŠĘų╬÷
ę“DBA╗╝ā║ČÓ▒Ē¼F(xi©żn)×ķ╠źā║įņč¬╠žš„Ż¼6éĆ(g©©)į┬ā╚(n©©i)č¬╝tĄ░░ūFŻ©hemoglobin FŻ¼HbFŻ®░┘Ęų▒╚ę▓▒╚š²│Ż═¼²gā║╔²Ė▀Ż¼6éĆ(g©©)į┬║¾HbF╚į│ų└m(x©┤)╔²Ė▀Ż¼ČÓ▒Ż│ųį┌5ŻźĪ½10ŻźŻ¼▀@ą®╠źā║śė╝t╝Ü(x©¼)░¹╠žš„▒Ē├„æ¬(y©®ng)╝żąį╝t╝Ü(x©¼)░¹╔·│╔ĪŻ
3.╝t╝Ü(x©¼)░¹ųąŽ┘▄š├ō░▒├ĖŻ©erythrocyte adenosine deaminaseŻ¼eADAŻ®╗Ņąį
╗╝š▀═©│Ż┤µį┌Óč▀╩║╦▄š┤·ųx«É│ŻŻ¼┐╔▒Ē¼F(xi©żn)×ķ47ŻźĪ½100Żź╗╝š▀│÷¼F(xi©żn)╝t╝Ü(x©¼)░¹Ž┘▄š├ō░▒├ĖĄ─╗Ņąį╔²Ė▀ĪŻ
4.╣Ū╦ĶÖz▓ķ
╠ß╩Š╣Ū╦Ķįņč¬ĮM┐Ś▒╚└²š²│Ż░ķ╝tŽĄŪ░¾w╝Ü(x©¼)░¹£p╔┘╗“╚▒╩¦ĪŻ90Żźęį╔Ž╗╝š▀Ą─╣Ū╦Ķį÷╔·│╠Č╚š²│ŻŻ¼āHįńŲ┌╝tŽĄ╝Ü(x©¼)░¹├„’@£p╔┘╗“╚▒╚ńŻ©ČÓŻ╝5ŻźŻ®Ż¼┴Ż╝t▒╚└²┐╔▀_(d©ó)10:1Ż¼┴Ż╝Ü(x©¼)░¹ŽĄ║═Š▐║╦╝Ü(x©¼)░¹ŽĄį÷╔·š²│ŻĪŻ
5.Ųõ╦¹Öz▓ķ
═Ų╦]▀M(j©¼n)ąąą─┼K│¼┬Ģ║═─I┼Kė░Ž±Öz▓ķ║Y▓ķŲ„╣┘░l(f©Ī)ė²«É│Ż┐╔─▄ĪŻ
į\öÓŻ║
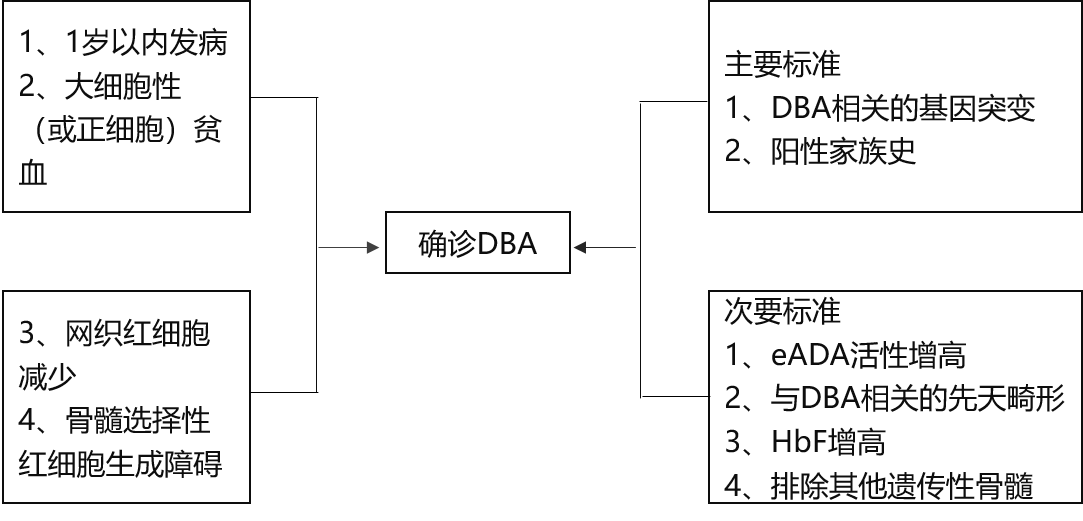
Ę¹║ŽęįŽ┬4Ślś╦(bi©Īo)£╩(zh©│n)Ą─╗╝š▀┐╔ęįį\öÓDBAŻ║
1.░l(f©Ī)▓Ī─Ļ²gąĪė┌1ÜqĪŻ
2.┤¾╝Ü(x©¼)░¹ąįŻ©╗“š²╝Ü(x©¼)░¹ąįŻ®žÜ謯¼░ū╝Ü(x©¼)░¹š²│Ż╗“╔įĮĄĄ═Īó謹Ī░Õš²│Ż╗“╔įį÷Ė▀ĪŻ
3.ŠW(w©Żng)┐Ś╝t╝Ü(x©¼)░¹├„’@£p╔┘ĪŻ
4.╣Ū╦Ķį÷╔·╗Ņ▄S░ķ╝tŽĄŪ░¾w╝Ü(x©¼)░¹├„’@£p╔┘ĪŻ
ī”(du©¼)ė┌▓╗ØMūŃęį╔Ž╦∙ėąį\öÓś╦(bi©Īo)£╩(zh©│n)Ą─╗╝š▀Ż¼┐╔į÷╝ėų¦│ųś╦(bi©Īo)£╩(zh©│n)Ż¼Ęų×ķų„ę¬║═┤╬ę¬░l(f©Ī)¼F(xi©żn)ęįū÷│÷Ī░öMį\Ī▒ĪŻ
1.ų„ę¬ų¦│ųś╦(bi©Īo)£╩(zh©│n)Ż║░³└©┤µį┌┼cDBAŽÓĻP(gu©Īn)Ą─╗∙ę“═╗ūā║═Ļ¢(y©óng)ąį╝ęūÕ╩ĘĪŻ
2.┤╬ę¬ų¦│ųś╦(bi©Īo)£╩(zh©│n)Ż║░³└©eADA╗Ņąįį÷Ė▀Īó┼cDBAŽÓĻP(gu©Īn)Ą─Ž╚╠ņ╗¹ą╬ĪóHbFį÷Ė▀║═┼┼│²Ųõ╦¹▀zé„ąį╣Ū╦Ķ╦źĮ▀ŠC║Žš„Ą─ūCō■(j©┤)ĪŻ
į┌ęįŽ┬ŪķørŽ┬┐╔öMį\DBAŻ║
1.ØMūŃ3ĒŚ(xi©żng)į\öÓś╦(bi©Īo)£╩(zh©│n)▓óėąĻ¢(y©óng)ąį╝ęūÕ╩ĘĪŻ
2.ØMūŃ2ĒŚ(xi©żng)į\öÓś╦(bi©Īo)£╩(zh©│n)║═3ĒŚ(xi©żng)┤╬꬜╦(bi©Īo)£╩(zh©│n)ĪŻ
2.Ļ¢(y©óng)ąį╝ęūÕ╩Ę║═3ĒŚ(xi©żng)┤╬꬜╦(bi©Īo)£╩(zh©│n)ĪŻ
ī”(du©¼)ė┌┼R┤▓Ė▀Č╚æčę╔DBAĄ─╗╝š▀Ż¼ę▓Į©ūh▀M(j©¼n)ąą┼cDBAŽÓĻP(gu©Īn)╗∙ę“═╗ūāĄ─╗∙ę“║Y▓ķĪŻūŅėąą¦Ą─ĘĮĘ©╩Ū╩ūŽ╚▀M(j©¼n)ąąRPS19 ╗∙ę“═╗ūāĄ─ą“┴ąĘų╬÷Ż©ę“?y©żn)ķįō╗∙ę“═╗ūā╩ŪDBAūŅ│ŻęŖ(ji©żn)Ą─▓Īę“Ż®Ż¼╚╗║¾═©▀^(gu©░)ĘųūėīW(xu©”)Öz£y(c©©)ĘĮĘ©Öz£y(c©©)Ųõ╦¹8éĆ(g©©)┼cDBAŽÓĻP(gu©Īn)Ą─╗∙ę“ĪŻ┤╦═ŌŻ¼╚ń╣¹╗╝š▀Š▀ėąDBAŽÓĻP(gu©Īn)Ą─╗∙ę“═╗ūāŻ¼Ą½▓╗ØMūŃDBAĄ─į\öÓś╦(bi©Īo)£╩(zh©│n)Ż¼ät┐╔į\öÓ×ķĘŪĄõą═DBAĪŻ
«a(ch©Żn)Ū░į\öÓŻ║40ŻźĪ½50ŻźĄ─DBA╗╝š▀×ķ│Ż╚Š╔½¾w’@ąį▀zé„Ż¼ŲõėÓ×ķ╔ó░l(f©Ī)Ą─╗“š▀ėą▓╗═¼▀zé„╠žš„Ą─╝ęūÕąįĪŻę“?y©żn)ķ╩▄Ž┬ĮĄĄ─═Ō’@┬╩╝░▌pųžā╔ĘNą╬╩Į╣▓┤µį┌═¼ę╗╝ęūVā╚(n©©i)Ą╚ę“╦žĄ─ė░ĒæŻ¼DBAĄ─▀zé„ęÄ(gu©®)┬╔║▄ļyŅA(y©┤)£y(c©©)Ż¼╝┤╩▄ČÓę“╦žė░ĒæŻ¼▒Ē¼F(xi©żn)ą═║═╗∙ę“ą═▓╗ę╗ų┬ĪŻį┌DBA╝ęūÕųąæ¬(y©®ng)▀M(j©¼n)ąą▀zé„ū╔įāųĖī¦(d©Żo)ā×(y©Łu)╔·ā×(y©Łu)ė²ĪŻ
Ķbäeį\öÓŻ║
1.ā║═»Ų┌Ģ║Ģr(sh©¬)ąįėū╝t╝Ü(x©¼)░¹£p╔┘░YŻ©transient erythroblastopenia of childhoodŻ¼TECŻ®
TEC╩Ūā║═»╝t╝Ü(x©¼)░¹╔·│╔£p╔┘ūŅ│ŻęŖ(ji©żn)Ą─▓Īę“Ż¼═©│Ż╩Ūė╔╬óąĪ▓ĪČŠB19Ėą╚Šę²ŲĄ─½@Ą├ąįČ╠Ģ║Ą─╝t╝Ü(x©¼)░¹╔·│╔▓╗┴╝ĪŻTEC▒Ē¼F(xi©żn)×ķę╗▀^(gu©░)ąįĄ─ūį╔Ē├Ōę▀Įķī¦(d©Żo)Ą─╝▓▓ĪŻ¼▓Ī│╠│╩ūįŽ▐ąįŻ¼ČÓ░l(f©Ī)╔·ė┌1Üqęį║¾Ż¼¤o(w©▓)Ļ¢(y©óng)ąį╝ęūÕ╩Ę╗“Ž╚╠ņ╗¹ą╬Ż¼ė┌░l(f©Ī)▓Ī║¾1Ī½2éĆ(g©©)į┬ā╚(n©©i)╗ųÅ═(f©┤)Ż¼ŅA(y©┤)║¾┴╝║├ĪŻš²╝Ü(x©¼)░¹ąįžÜč¬Īóč¬╝tĄ░░ūļŖėŠ▀M(j©¼n)ąąHbF║¼┴┐║═eADAš²│ŻĪó╗∙ę“Ęų╬÷ėąų·ė┌ĶbäeTEC║═DBAĪŻ
2.AaseŠC║Žš„
╩Ūęį╔Žų½╗¹ą╬║Ž▓óŽ╚╠ņąį╝tŽĄ╔·│╔šŽĄK×ķų„ę¬▒Ē¼F(xi©żn)Ą─ŠC║Žš„ĪŻ╩▄└█ā║═»ę▓┐╔─▄ėąą─č¬╣▄║═’B├µ▓┐Ż©┤Įļ±┴诮╗¹ą╬ĪŻ┤╦ŠC║Žš„║▄┐╔─▄╩ŪDBAĄ─ę╗ĘNūā«Éą═Č°▓╗╩Ūę╗ĘN¬Ü(d©▓)┴óĄ─┼R┤▓╝▓▓ĪĪŻ┼cDBAę╗śėŻ¼ę╗▓┐Ęų╗╝š▀╩╣ė├Ųż┘|(zh©¼)ŅÉ╣╠┤╝ėąą¦ĪŻ
3.Ųõ╦¹Ž╚╠ņąį╣Ū╦Ķ╦źĮ▀ąį╝▓▓Ī
░³└©ĘČ┐Ų─ßžÜč¬ĪóShwachman-DiamondŠC║Žš„║═Ž╚╠ņąįĮŪ╗»▓╗┴╝ĪŻ┤╦ŅÉ╝▓▓Ī│Ż░ķėąŅ~═ŌĄ─č¬╝Ü(x©¼)░¹£p╔┘Ż¼ę“▒Ēą═Å═(f©┤)ļsČÓūāŻ¼╗∙ę“▌oų·┼R┤▓į\öÓĖ³×ķų▒ĮėĪŻ
╚²Īóų╬»¤ĘĮ╩Į
ų╬»¤Ż║DBAĄ─ų„ę¬ų╬»¤×ķŲż┘|(zh©¼)ŅÉ╣╠┤╝║═▌öč¬ĪŻ┐é¾w╔ŽŻ¼┤¾╝s40ŻźĄ─DBA╗╝š▀×ķŅÉ╣╠┤╝ę└┘ćą═Ż¼40Żź×ķ▌öč¬ę└┘ćą═Ż¼20ŻźĄĮ25ÜqĢr(sh©¬)┐╔½@Ą├ĄĮŠÅĮŌĪŻ10ŻźĪ½25Żź╗╝š▀┐╔ūį░l(f©Ī)ŠÅĮŌŻ╗╝s70ŻźĮø(j©®ng)ų╬»¤┐╔▀_(d©ó)═Ļ╚½ŠÅĮŌŻ¼Ą½╚įėą▓┐Ęų╗╝š▀Å═(f©┤)░l(f©Ī)Ż╗▓┐Ęų╗╝š▀ų╬»¤ą¦╣¹▌^▓ŅŻ¼ų„ę¬┐┐▌öč¬Ė─╔Ų░YĀŅŻ¼╣╩ęūę²Ųč¬╔½▓ĪĄ╚ĪŻ▓┐Ęųą¦╣¹ėąŽ▐Ą─ų╬»¤▀xō±ł¾(b©żo)Ą└Ż¼╚ń░ūĮķ╦ž-3Īóą█╝ż╦žĪó╝ūč§┬╚Ųš░ĘĪó├Ōę▀ęųųŲä®╚ńŁh(hu©ón)µ▀╦žĄ╚ĪŻ
1.╠ŪŲż┘|(zh©¼)╝ż╦ž
│Żė├ØŖ─ß╦╔2mg/(kgĪżd)Ż¼ęįįń│┐å╬ä®┴┐ę╗┤╬╗“Ęųā╔┤╬Įo╦ÄĪŻ50ŻźĪ½70Żź╗╝š▀│§┤╬ų╬»¤Ģr(sh©¬)ėąą¦ĪŻįĮįńų╬»¤Ż¼ėąą¦┬╩įĮĖ▀Ż¼Į©ūh▒MįńĮė╩▄ų╬»¤ĪŻų╬»¤ėąą¦Ą─╗╝š▀═©│Żį┌ų╬»¤║¾1Ī½2ų▄╝┤┐╔│÷¼F(xi©żn)ŠW(w©Żng)┐Ś╝t╝Ü(x©¼)░¹▒╚└²Ą─╔²Ė▀ĪŻHb╔Ž╔²ų┴100g/Lęį╔Ž║¾┐╔ķ_(k©Īi)╩╝£p┴┐ĪŻ8Ī½12ų▄ā╚(n©©i)æ¬(y©®ng)ŠÅ┬²£p╔┘╝ż╦žä®┴┐ų▒ĄĮ▀_(d©ó)ĄĮūŅąĪėąą¦ä®┴┐ĪŻ▀@ĘNūŅĄ═ėąą¦ä®┴┐ūā╗»║▄┤¾Ż¼▓óŪęįSČÓī”(du©¼)ŅÉ╣╠┤╝ėąĘ┤æ¬(y©®ng)Ą─╗╝ā║▓╗─▄═Ļ╚½═Ż╦ÄŻ¼ųØu£p┴┐─┐ś╦(bi©Īo)×ķ▀_(d©ó)ĄĮä®┴┐Ī▄0.5mg/(kgĪżd)Ż¼─┐ś╦(bi©Īo)č¬╝tĄ░░ū×ķ80Ī½100g/LĪŻ╗╝š▀╚įį┌╩╣ė├ųąĄ╚ęį╔Žä®┴┐╝ż╦žų╬»¤Ģr(sh©¬)Ż¼æ¬(y©®ng)▒▄├Ō╩╣ė├╗Ņ▓ĪČŠę▀├ńĪŻų╬»¤æ¬(y©®ng)ćLįć▀M(j©¼n)ąą4ų▄Ż¼╚ń╣¹¤o(w©▓)Ę┤æ¬(y©®ng)Ż¼ätæ¬(y©®ng)ųąų╣ų╬»¤ĪŻŅÉ╣╠┤╝¤o(w©▓)ą¦š▀ąĶŠ»╠ĶŽ“╣Ū╦Ķ╦źĮ▀░l(f©Ī)š╣Ż¼Š▀ėąRPS26 ╗∙ę“═╗ūāĄ─╗╝š▀ī”(du©¼)ŅÉ╣╠┤╝ų╬»¤Ą─Ę┤æ¬(y©®ng)┬╩ūŅĄ═ĪŻ
ąĶę¬ūóęŌŻ║į┌ŗļā║ųą╩╣ė├ŅÉ╣╠┤╝ėąć└(y©ón)ųžĄ─▓╗┴╝Ę┤æ¬(y©®ng)Ż¼ė╚Ųõ╩Ūįń«a(ch©Żn)ā║ųą╝ż╦žķL(zh©Żng)Ų┌æ¬(y©®ng)ė├┐╔─▄│÷¼F(xi©żn)╔·ķL(zh©Żng)čė▀tŻ¼īW(xu©”)▓ĮĢr(sh©¬)Ų┌ęū│÷¼F(xi©żn)╔±Įø(j©®ng)╝Ī╚Ō░l(f©Ī)ė²▓╗┴╝╝░▀\(y©┤n)äė(d©░ng)╔±Įø(j©®ng)čė▀tĪŻ╦∙ęįČÓöĄ(sh©┤)īŻ╝ę═Ų╦]į┌6Ī½12į┬²gų«Ū░æ¬(y©®ng)▒▄├Ō╩╣ė├ŅÉ╣╠┤╝ų╬»¤Ż¼į┌┤╦Ų┌ķg┐╔ęį═©▀^(gu©░)▌öč¬ų╬»¤▀M(j©¼n)ąą╠µ┤·ĪŻī”(du©¼)ė┌ķL(zh©Żng)Ų┌Įė╩▄ØŖ─ß╦╔ų╬»¤Ą─╗╝š▀æ¬(y©®ng)┐╝æ]ķ_(k©Īi)╩╝ŅA(y©┤)Ę└ąį╩╣ė├╗Ū░Ę╝ūÉ║▀“-╝ūč§ŲSÓżų╬»¤ĪŻ
2.▌öč¬ų╬»¤
ī”(du©¼)ė┌Ųż┘|(zh©¼)ŅÉ╣╠┤╝ų╬»¤¤o(w©▓)ą¦╗“┤µį┌ŅÉ╣╠┤╝╩╣ė├Į¹╝╔Ą─╗╝š▀Ż¼▌öč¬╩Ūų„ę¬ų╬»¤╩ųČ╬ĪŻ┤╦ŅÉ╗╝š▀ę└┘ćķL(zh©Żng)Ų┌ķgöÓąį▌ö謯¼æ¬(y©®ng)▀M(j©¼n)ąą═Ļ╚½╝t╝Ü(x©¼)░¹Ęųą═Īó╚ź│²░ū╝Ü(x©¼)░¹╠Ä└ĒŻ¼▒▄├Ōų▒ŽĄėHī┘╣®ĮoĪŻ═©│Ż├┐4Ī½6ų▄▌öč¬1┤╬Ż¼č¬╝tĄ░░ū╦«ŲĮŠS│ųį┌80g/Lęį╔ŽŻ¼┐╔▒ŻūC╔·ķL(zh©Żng)░l(f©Ī)ė²╝░╚š│Ż╗Ņäė(d©░ng)ąĶę¬ĪŻ
ķL(zh©Żng)Ų┌▌ö謥─╗╝ā║┐╔ę²ŲĶF▀^(gu©░)▌dŻ¼æ¬(y©®ng)▒O(ji©Īn)£y(c©©)č¬ŪÕĶFĄ░░ūŻ¼╝░Ģr(sh©¬)╚źĶFų╬»¤Ż¼Į©ūh╚źĶFų╬»¤┐╔ęįį┌▌öūóæęĖĪ╝t╝Ü(x©¼)░¹15┤╬║¾╗“╩Ū╗╝ā║ØM2Üq║¾ķ_(k©Īi)╩╝ĪŻ═Ų╦]Ąž└Ł┴_╦Š20mg/(kgĪżd)Ų╩╝Ż¼ŠÅ┬²╝ė┴┐Ż¼ūŅ┤¾ä®┴┐▓╗│¼▀^(gu©░)40mg/(kgĪżd)ĪŻ├┐3éĆ(g©©)į┬▒O(ji©Īn)£y(c©©)č¬ŪÕĶFĄ░░ūŻ¼─┐ś╦(bi©Īo)ųĄ×ķ1000Ī½1500”╠g/LĪŻ
3.╗∙ę“ų╬»¤
░ķėąRPS19 ╗∙ę“╚▒Ž▌Ą─DBA╗╝š▀Ą─╗∙ę“ų╬»¤š²į┌蹊┐ųąŻ¼¾w═ŌīŹ(sh©¬)“×(y©żn)’@╩ŠŻ¼į┌╗╝š▀╝tŽĄūµ╝Ü(x©¼)░¹ųąį÷╝ėRPS19 ╗∙ę“Ą─▒Ē▀_(d©ó)┐╔┤┘▀M(j©¼n)╝t╝Ü(x©¼)░¹░l(f©Ī)ė²Ż¼ė├▐D(zhu©Żn)╗∙ę“▓ĪČŠ▌d¾wüĒ(l©ói)“×(y©żn)ūC╗∙ę“ų╬»¤╣”ą¦Ą─äė(d©░ng)╬’─Żą═Ż©╚ź│²RPS19 ╗∙ę“▒Ē▀_(d©ó)Ż®ęčĮø(j©®ng)▒╗│╔╣”ųŲ│╔ĪŻ
4.įņč¬Ė╔╝Ü(x©¼)░¹ęŲų▓
ŅÉ╣╠┤╝¤o(w©▓)ą¦Īó▌öč¬ę└┘ćą═DBA┐╔ęį┐╝æ]įņč¬Ė╔╝Ü(x©¼)░¹ęŲų▓ĪŻ═¼░¹╣®š▀ąĶūóęŌ║Y▓ķ┼┼│²╣®š▀öyĦų┬▓Ī╗∙ę“═╗ūāĪŻ
į\»¤┴„│╠łDŻ║
Ž╚╠ņąį╝ā╝t╝Ü(x©¼)░¹į┘╔·šŽĄKąįžÜč¬į\»¤┴„│╠
▀zé„ū╔įāŻ║40ŻźĪ½50ŻźĄ─DBA╗╝š▀×ķ│Ż╚Š╔½¾w’@ąį▀zé„Ż¼ŲõėÓ×ķ╔ó░l(f©Ī)Ą─╗“š▀ėą▓╗═¼▀zé„╠žš„Ą─╝ęūÕąįĪŻę“?y©żn)ķ╩▄Ž┬ĮĄĄ─═Ō’@┬╩╝░▌pųžā╔ĘNą╬╩Į╣▓┤µį┌═¼ę╗╝ęūVā╚(n©©i)Ą╚ę“╦žĄ─ė░ĒæŻ¼DBAĄ─▀zé„ęÄ(gu©®)┬╔║▄ļyŅA(y©┤)£y(c©©)Ż¼╝┤╩▄ČÓę“╦žė░ĒæŻ¼▒Ē¼F(xi©żn)ą═║═╗∙ę“ą═▓╗ę╗ų┬ĪŻį┌DBA╝ęūÕųąæ¬(y©®ng)▀M(j©¼n)ąą▀zé„ū╔įāųĖī¦(d©Żo)ā×(y©Łu)╔·ā×(y©Łu)ė²ĪŻ
ģó┐╝╬─½I(xi©żn)Ż║
[1] Åłų«─Ž,║┬ė±Ģ°,┌wė└ÅŖ(qi©óng),═§Į©ŽķĄ╚.č¬ę║▓ĪīW(xu©”).Ą┌2░µ.▒▒Š®:╚╦├±ąl(w©©i)╔·│÷░µ╔ń,2011:484-485.
[2] Willig TN, Gazda H, Sieff CA. Diamond-Blackfan anemia. Curr Opin Hematol,2000,7:85.
[3] Vlachos A, Ball S, Dahl N, et al. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol,2008,142:859.
[4] Vlachos A, Rosenberg PS, Atsidaftos E, et al. Incidence of neoplasia in Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Blood,2012,119:3815.
[5] Halperin DS, Freedman MH. Diamond-blackfan anemia: etiology, pathophysiology, and treatment. Am J Pediatr Hematol Oncol, 1989,11:380.
[6] Farrar JE, Nater M, Caywood E, et al. Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond-Blackfan anemia. Blood, 2008,112:1582.
[7] Orfali KA, Ohene-Abuakwa Y, Ball SE. Diamond Blackfan anaemia in the UK: clinical and genetic heterogeneity. Br J Haematol, 2004, 125:243.
[8] Kuramitsu M, Sato-Otsubo A, Morio T, et al. Extensive gene deletions in Japanese patients with Diamond-Blackfan anemia. Blood, 2012, 119:2376.
[9] Chen S, Warszawski J, Bader-Meunier B, et al. Diamond-Blackfan anemia and growth status: the French registry. J Pediatr, 2005, 147:669.
[10] Abkowitz JL, Schaison G, Boulad F, et al.Response of Diamond-Blackfan anemia to metoclopramide: evidence for a role for prolactin in erythropoiesis. Blood, 2002,100(8):2687.
[11] Ball SE, Tchernia G, Wranne L, et al. Is there a role for interleukin-3 in Diamond-Blackfan anaemia? Results of a European multicentre study. Br J Haematol,1995,91(2):313.
[12] Leonard EM, Raefsky E, Griffith P, et al. Cyclosporine therapy of aplastic anaemia, congenital and acquired red cell aplasia. Br J Haematol,1989,72(2):278.
░µÖÓ(qu©ón)┬Ģ├„
ęį╔Žā╚(n©©i)╚▌üĒ(l©ói)ūį┴╝ßt(y©®)ģR-║▒ęŖ(ji©żn)▓Īą┬▀M(j©¼n)š╣Ż¼╚ńėąĮ©ūh╗“ę╔å¢(w©©n)Ż¼ÜgėŁų┬ļŖ18017449015ĪŻ
 ķL(zh©Żng)░┤Č■ŠS┤a
ķL(zh©Żng)░┤Č■ŠS┤aĻP(gu©Īn)ūóŠ½▓╩ā╚(n©©i)╚▌





